

Simulating liquid electrolytes on a computer enables electrochemists to understand the intricate details of liquid electrolyte structure and the underlying causes of phenomena that occur within them, with the ultimate goal of utilizing this knowledge to design efficient electrochemical devices. Moreover, it allows us to predict the properties of electrolytes that have not yet been experimentally studied, which is important for the targeted design of electrochemical devices. To simulate electrolytes, we use a variety of approaches: (1) quantum chemical modeling of electrolyte components; (2) molecular dynamics simulation using empirical force fields; (3) machine learning methods to predict the physical properties of electrolyte solution components. Below are examples of the research we have done.
Quantum-Chemical Studies and Molecular Dynamics Simulations of Liquid Electrolyte in Rechargeable Silicon RedOx Batteries
The demand for energy storage has led to the development of side-by-side technologies that can supplement the current Lithium-ion battery industry with more affordable and abundant materials. Silicon, the second most abundant element in the Earth’s crust, has made promising progress in the last decade, and recent advances in Silicon RedOx battery design indicate a promising path forward. The use of tetra-butyl ammonium fluoride trihydrate (TBAF · 3H2O) dissolved in organic solvents with a wide electrochemical window as an electrolyte enables efficient surface activation and satisfactory electrolyte conductivity while minimizing the hydrogen evolution reaction.
To deeply understand the factors influencing the internal structure, spectroscopic, physical, and transport properties of the electrolyte components, we undertook quantum-chemical density functional theory (DFT) modeling of the species that make up the electrolyte, their interactions with each other, as well as empirical force-fields-based molecular dynamics simulations of electrolytes with different organic solvents and their mixtures and different concentrations of the dissolved salt (TBAF∙3H2O). The influence of the physical characteristics of the solvent (dielectric constant, acidity/basicity) on fluoride specification, solvation, and electrolyte performance, as well as prediction of 19F NMR shifts, ionic conductivity mechanisms, and fluoride reactivity, was the focus of the study. The results obtained helped to design Silicon RedOx batteries with fluoride-based electrolytes with optimal ionic conductivity, adequate anode surface activation, current density, and electrochemical stability with corresponding cell characterization and operation mechanism.
Dependence of Reaction Free Energy on Dielectric Constant ɛ
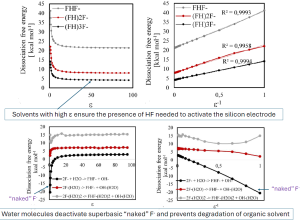
We have evaluated the propensity of the fluoride anion to form complexes with HF molecules in the gas phase and several solutions using quantum-chemical DFT calculations using continuum solvation models. The complexation free energy was estimated as the difference between the formation free energy of the fluorohydrogenate anion and the sum of the formation free energies of its components (F– and HF). The effect of adding each subsequent HF molecule was shown to decrease as n increases. Although the absolute complexation free energy in vacuum increases significantly with the addition of each HF molecule, in solutions, the addition of the third HF molecule gives a significantly smaller gain in free energy. Therefore, it can be expected that a simultaneous presence of comparable amounts of (FH)2F– and (FH)3F– could be found in the solution, as observed in the experiment. It has also been shown that the free dissociation energy of HF decreases significantly with an increase in the number of HF units in the complex, as well as when moving from the gas phase to a solvent. This explains several experimental observations. In particular, since two reversible processes of HF cleavage from these two complexes and their reverse binding are quite sufficient for a very fast mutual exchange of terminal and central fluorine atoms even at low temperatures, as supported by 19F NMR spectra. As follows from the calculations, very low free dissociation energies lead to high ionic conductivity and can be theoretically predicted by solely including interaction with the polarizable medium in quantum chemical calculations.
The concept of a “naked” fluoride anion comes from synthetic organic chemistry and denotes a fluoride anion in aprotic solvents, with which it does not form hydrogen bonds or coordination bonds, forming its “clothes” and is therefore called “naked”, and acts as a very strong base and nucleophile. However, its basicity and
nucleophilicity can be significantly reduced in protic solvents by forming hydrogen bonds with the fluoride anion to yield oligofluorinated species. Herein, we applied computational methodology as discussed above to model and explain this phenomenon. We have also explained the reason why the addition of even a small amount of water to a solution of a fluoride salt in an aprotic solvent can lead to the same deactivation of the “naked” anion as substitution to a protic solvent i. e. water; hence, we can enjoy high solubility, conductivity and stability with minimal concentration of water whilst mitigating its adverse effects on electrolyte stability.
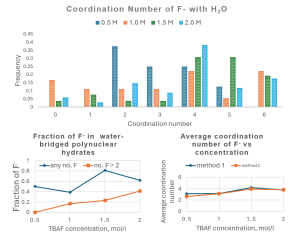
Simulation of Electrolyte. Effect of F- Bridging via Water Molecules
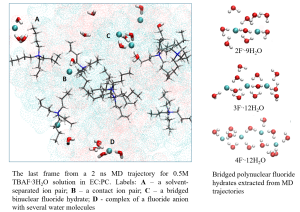
Details of F- Coordination at Different Concentrations of TBAF Extracted from MD Trajectories
Molecular Dynamics Simulation of Talc Nanosheet Ionogel Electrolyte with High Lithium-Ion Conductivity for Solid-State Lithium-Ion Batteries
Ionogels are an appealing solid-state electrolyte for lithium-ion batteries due to their nonflammability, good electrochemical characteristics, and extensive manufacturing compatibility. However, they often have poor lithium-ion conductivity due to low lithium-ion transference numbers, which limits their battery performance. We have discovered ionogel electrolytes with high lithium-ion conductivity based on liquid-phase-exfoliated talc nanosheets and an imidazolium ionic liquid. To explain the phenomenon of a sharp increase in the ionic conductivity of a lithium salt solution in an ionic liquid upon the addition of talc nanosheets, molecular dynamics simulations were performed. They have revealed that talc nanosheets reduce the coordination of lithium cations with ionic liquid anions and induce the formation of less extended and looser complexes, thus promoting higher ionic conductivity.
Talc nanosheet with ionic liquid
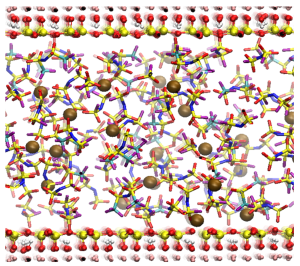
Snapshot of the MD trajectory for the Li-IL with talc nanosheets. EMIM cations are omitted
MD simulations were performed for two Li-IL systems of 1 M LiTFSI/EMIM-FSI with and without talc nanosheets to elucidate the influences of the talc nanosheets..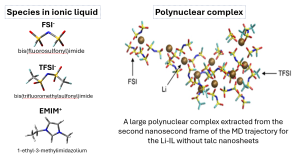
Simulation results reveal that lithium ions in the Li-IL tend to form polynuclear complexes with polydentate FSI and TFSI anions, which create bridges that hold the lithium ions together. The Figure above shows a snapshot of a large polynuclear complex extracted from the MD trajectory for the Li-IL without talc nanosheets, containing 11 lithium cations glued together by bridges formed from 23 FSI and 3 TFSI anions. The complex is also surrounded by a shell of EMIM cations, which is not shown in the snapshot. The complex formation occurs because each of these anions contains several oxygen atoms in its sulfonyl groups, which have a high partial negative charge and are thus prone to complexation with lithium cations. As a result, the polynuclear complexes form based on Li+…O=S=O…Li+ bridges.
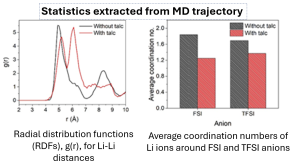
MD simulations reveal that talc nanosheets reduce the coordination of lithium cations with ionic liquid anions, inducing the formation of less extended and looser complexes. This significantly improves lithium-ion transference numbers.
Read more details in the paper.
Benchmarking machine learning methods for modeling physical properties of ionic liquids
Ionic liquids (ILs) possess a unique combination of physicochemical properties that make them extremely important in the development of clean and sustainable liquid electrolyte materials for electrochemical energy storage and conversion devices, especially as electrolytes in Li-ion, Na-ion, Li-air, Li-S, all-solid-state, dual-ion, and other types of batteries, as well as supercapacitors. The importance of the ability to perform quantitative predictions of various properties of ILs using quantitative structure-property relationships (QSPR) models necessitates the understanding of which modern machine learning (ML) methods, in combination with which types of molecular descriptors, are preferable to use for this purpose.
We have performed a large-scale benchmarking study of QSPR models built by combining three traditional ML methods and seven different architectures of neural networks (NNs) with five types of molecular descriptors to predict six important physical properties of ILs: density, electrical conductivity, melting point, refractive index, surface tension, and viscosity [3]. The models are built using datasets consisting of 407 to 1204 ILs composed of diverse organic and inorganic ions. QSPR models for predicting the properties of ILs at eight different temperatures are built using multi-task learning. For each of the properties, the best combinations of ML methods and molecular descriptors are identified. A unified ranking system is used to rank and prioritize different ML methods and molecular descriptors. It is shown that on average: (1) nonlinear ML methods perform much better than linear ones; (2) NNs perform better than traditional ML methods; (3) NNs with attention-based Transformer architectures, which are actively used in natural language processing (NLP) and other fields of artificial intelligence (AI), perform better than other types of NNs due to the advanced ability to analyze SMILES text strings encoding chemical structures of ILs. The resulting models can be applied both to predict the properties of new ILs and to design ILs with the desired set of physical properties.